Last updated: June 27, 2026
The European Union Medical Device Regulation (EU MDR 2017/745) fundamentally changed how nitrile gloves are classified, tracked, and sold across European markets. Under EU MDR, medical-grade nitrile gloves require Unique Device Identification (UDI) codes, comprehensive technical documentation, and rigorous supplier vetting processes that distributors and manufacturers must implement to maintain market access. These traceability requirements and supplier vetting protocols directly impact procurement costs, supply chain management, and compliance risk for healthcare facilities, distributors, and industrial buyers sourcing nitrile gloves for medical applications.
Key Takeaways
- EU MDR classifies medical nitrile gloves as Class I medical devices requiring UDI codes, technical files, and authorized representative designation for non-EU manufacturers
- Traceability requirements mandate batch-level tracking with UDI-DI (Device Identifier) and UDI-PI (Production Identifier) on all medical glove packaging
- Supplier vetting must verify ISO 13485 certification, EU MDR technical documentation, and ongoing post-market surveillance capabilities
- Exam gloves and surgical gloves face different classification levels under EU MDR, with surgical gloves requiring stricter sterility validation
- Non-compliant nitrile gloves cannot legally be sold as medical devices in the EU after the May 2021 transition deadline (extended grace periods ended May 2024)
- Distributors share legal responsibility for ensuring their suppliers maintain valid EU MDR certification and proper documentation
- Compliance costs typically add 8-15% to nitrile glove procurement expenses through certification fees, documentation requirements, and enhanced quality control
- Common implementation mistakes include incomplete UDI database registration, inadequate supplier audit trails, and failure to maintain updated technical files
What Are the EU MDR Traceability Requirements for Nitrile Gloves
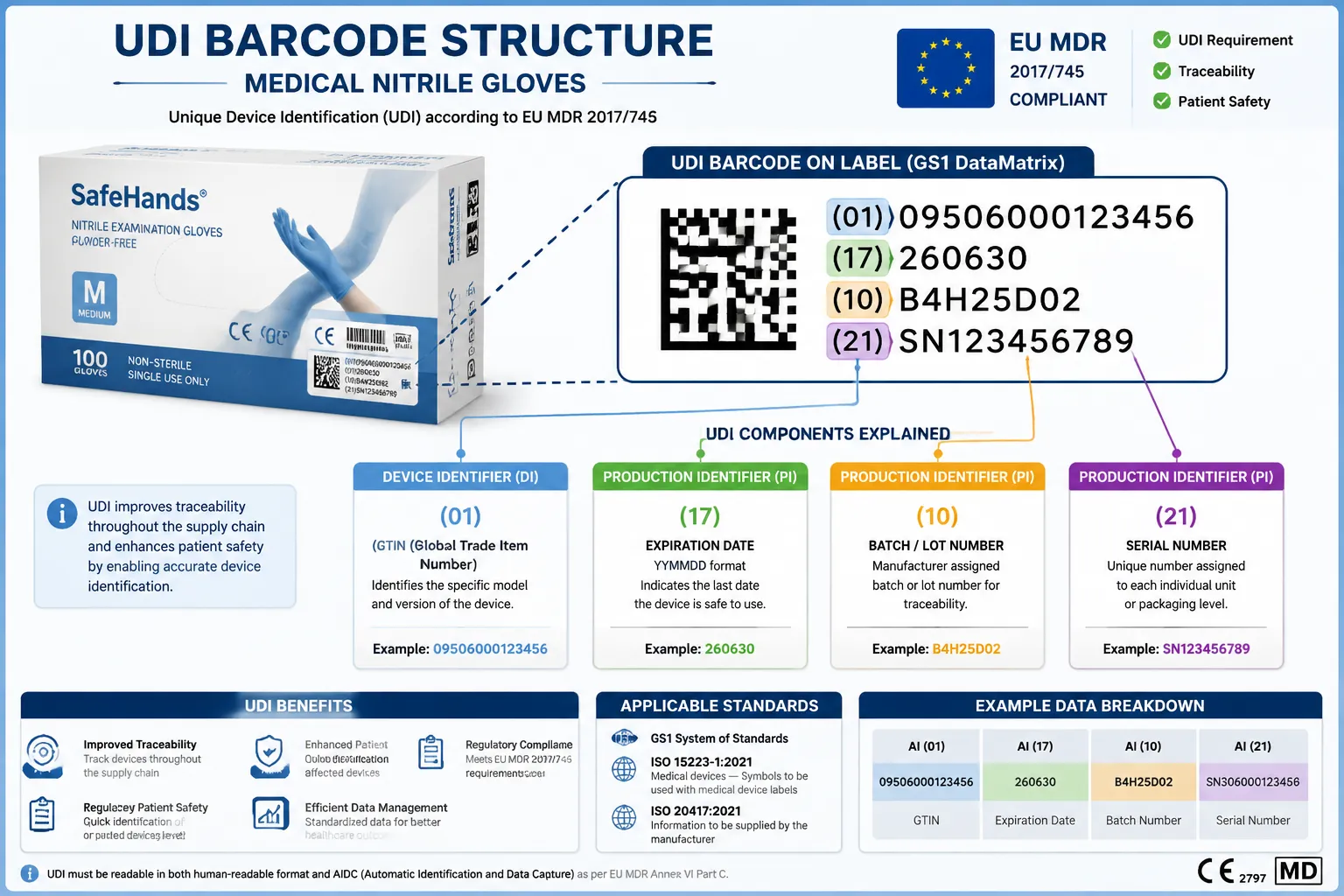
EU MDR traceability requirements for nitrile gloves mandate that every medical-grade glove product carry a Unique Device Identification (UDI) system that enables tracking from manufacturer to end user. The UDI consists of two components: a Device Identifier (UDI-DI) that identifies the specific glove model and manufacturer, and a Production Identifier (UDI-PI) that captures batch number, expiration date, and serial number when applicable.
Core traceability elements required under EU MDR:
- UDI-DI barcode printed on primary packaging (individual boxes)
- UDI-PI information including lot/batch number and expiration date
- Registration in the European Database on Medical Devices (EUDAMED)
- Economic operator identification for manufacturer, authorized representative, and importer
- Complete supply chain documentation from raw material sourcing through distribution
The regulation requires manufacturers to maintain traceability records for a minimum of 10 years after the last device has been placed on the market, or 15 years for implantable devices. For nitrile surgical gloves and exam gloves used in hospitals, this means comprehensive batch tracking systems that can quickly identify and recall specific production runs if quality issues arise.
Practical implementation steps:
- Obtain a UDI issuing entity code (GS1, HIBCC, or ICCBBA)
- Generate unique UDI-DI codes for each glove product variant
- Implement barcode printing on all packaging levels
- Register UDI data in EUDAMED database
- Establish internal systems linking UDI codes to production records
- Train staff on UDI scanning and documentation protocols
A common mistake is treating UDI implementation as a one-time labeling change rather than an ongoing data management system. Manufacturers must update EUDAMED entries whenever product specifications, packaging, or labeling changes occur.
Do Nitrile Gloves Need UDI Codes Under EU MDR
Yes, all nitrile gloves marketed as medical devices in the EU require UDI codes under EU MDR Article 27. This applies to examination gloves, surgical gloves, and any other nitrile gloves making medical claims or intended for patient contact in healthcare settings.
The UDI requirement applies at multiple packaging levels:
Primary packaging (individual boxes): Must display both UDI-DI and UDI-PI in human-readable format and machine-readable barcode (typically GS1-128 or Data Matrix).
Secondary packaging (case/carton level): Must include UDI information that corresponds to the primary packaging contents.
Tertiary packaging (pallet level): Should include aggregated UDI data for logistics tracking, though requirements are less stringent.
Industrial or non-medical nitrile gloves that make no medical claims and are not intended for patient examination or surgical procedures do not require UDI codes. However, the distinction must be clear in marketing materials and product labeling. If a glove is sold to healthcare facilities or marketed with terms like “exam grade” or “medical quality,” EU authorities will likely classify it as a medical device requiring full UDI compliance.
Choose UDI implementation based on:
- If selling to hospitals or clinics: Full UDI compliance mandatory
- If selling for industrial use only: UDI not required, but avoid medical terminology
- If selling both markets: Separate product lines with distinct SKUs and labeling
The transition period for UDI implementation ended in May 2024 for Class I devices, meaning all medical nitrile gloves currently on the EU market must carry compliant UDI codes.
How to Vet Suppliers for EU MDR Compliant Nitrile Gloves

Vetting suppliers for EU MDR compliant nitrile gloves requires verifying three critical documentation areas: quality management system certification, technical file completeness, and authorized representative designation. Start by requesting the supplier’s ISO 13485 certificate (current and unrevoked), EU MDR Declaration of Conformity, and proof of EUDAMED registration for their glove products.
Essential supplier vetting checklist:
- ISO 13485 certification: Verify certificate is current, covers glove manufacturing, and issued by a recognized certification body
- Declaration of Conformity: Confirm document references EU MDR 2017/745 (not old MDD directive) and lists specific glove models
- Technical documentation: Request summary of technical file including risk analysis, biocompatibility testing, and performance validation
- Authorized representative: For non-EU manufacturers, verify valid AR agreement with EU-based entity
- EUDAMED registration: Confirm manufacturer’s Single Registration Number (SRN) and product UDI-DI codes appear in database
- Post-market surveillance plan: Review supplier’s process for handling complaints, vigilance reporting, and corrective actions
- Supply chain transparency: Verify raw material sourcing documentation and subcontractor quality agreements
Request copies of recent third-party audit reports if available. Reputable suppliers will have undergone Notified Body assessments or independent quality audits that validate their EU MDR compliance systems.
Red flags during supplier vetting:
- Reluctance to provide ISO 13485 certificate or Declaration of Conformity
- Certificate issued by unknown or non-accredited certification body
- Declaration of Conformity still references MDD (Directive 93/42/EEC) instead of MDR
- No authorized representative listed for non-EU manufacturers
- UDI codes not registered in EUDAMED database
- Inability to provide batch-level traceability documentation
For ongoing supplier management, establish quarterly compliance reviews and require suppliers to notify you immediately of any certification changes, Notified Body audits, or regulatory non-conformances. Many distributors now include EU MDR compliance clauses in purchase agreements that allow contract termination if certification lapses.
Difference Between EU MDR and Old MDD for Medical Gloves
The EU Medical Device Directive (MDD 93/42/EEC) and EU Medical Device Regulation (MDR 2017/745) differ significantly in classification rigor, documentation requirements, and post-market surveillance obligations for medical gloves. Under MDD, manufacturers could self-certify Class I medical gloves with minimal oversight, while EU MDR requires comprehensive technical files, UDI implementation, and enhanced clinical evaluation even for basic examination gloves.
Key regulatory differences:
| Aspect | MDD (93/42/EEC) | EU MDR (2017/745) |
|---|---|---|
| Classification basis | Risk-based, minimal criteria | Enhanced risk classification with stricter definitions |
| Technical documentation | Basic technical file | Comprehensive technical documentation with clinical evaluation |
| UDI requirement | Not required | Mandatory for all medical devices |
| Post-market surveillance | Limited requirements | Systematic post-market surveillance plan mandatory |
| Economic operator responsibilities | Primarily manufacturer | Shared responsibility across distributors and importers |
| Notified Body involvement | Optional for Class I | Required for sterile/measuring function devices |
| Vigilance reporting | Basic incident reporting | Enhanced vigilance with serious incident timelines |
| EUDAMED registration | Not required | Mandatory registration for all devices |
The transition from MDD to MDR fundamentally changed the compliance burden for glove manufacturers. Under MDD, a manufacturer could create a basic technical file, issue a Declaration of Conformity, and affix CE marking with minimal external validation. EU MDR requires ongoing documentation updates, systematic literature reviews for biocompatibility, and detailed risk management files that must be maintained throughout the product lifecycle.
For exam gloves versus surgical gloves, MDR introduced clearer distinctions. Surgical gloves now explicitly require sterility validation and fall under stricter scrutiny, while examination gloves remain Class I but with significantly enhanced documentation expectations compared to MDD.
The practical impact: manufacturers who relied on minimal MDD compliance faced substantial investment in quality systems, documentation, and testing to meet MDR standards. Many smaller manufacturers exited the EU market rather than absorb these compliance costs.
Are All Nitrile Gloves Considered Medical Devices Under EU MDR
No, only nitrile gloves intended for medical purposes or making medical claims are classified as medical devices under EU MDR. The regulation defines medical devices based on intended purpose rather than product characteristics, so identical nitrile gloves can be medical devices or general-purpose protective equipment depending on how they are marketed and used.
Medical device classification applies when gloves are:
- Intended for patient examination or diagnosis
- Used in surgical or invasive procedures
- Marketed to healthcare facilities with medical terminology
- Labeled with claims about infection control or patient protection
- Designed to prevent cross-contamination in clinical settings
Non-medical device classification applies when gloves are:
- Marketed solely for industrial applications (automotive, manufacturing, food service)
- Labeled for general-purpose protection without medical claims
- Sold through non-medical distribution channels
- Intended for personal protective equipment (PPE) use under EU Regulation 2016/425
The distinction matters because medical device gloves require EU MDR compliance (UDI codes, technical files, Declaration of Conformity), while industrial gloves fall under PPE regulations with different testing and certification requirements. Some manufacturers produce identical glove formulations but maintain separate product lines with distinct labeling and distribution channels to serve both markets.
Gray area considerations:
Gloves sold to dental offices, veterinary clinics, or tattoo parlors may trigger medical device classification even if not explicitly marketed as medical. EU authorities evaluate intended purpose based on marketing materials, distribution channels, and reasonably foreseeable use. If a manufacturer knows their gloves are predominantly used in healthcare settings, claiming “industrial use only” may not exempt them from medical device regulations.
For nitrile gloves used in food handling, classification depends on whether infection control claims are made. Food-safe gloves without medical claims fall under food contact material regulations, not EU MDR.
What Documentation Do I Need From Nitrile Glove Suppliers for EU MDR
For EU MDR compliance, you need seven core documents from nitrile glove suppliers: Declaration of Conformity, ISO 13485 certificate, technical file summary, UDI registration confirmation, authorized representative agreement (for non-EU manufacturers), post-market surveillance plan, and batch release documentation. These documents establish the legal basis for placing medical gloves on the EU market and demonstrate ongoing compliance.
Essential supplier documentation:
Declaration of Conformity (DoC): Must reference EU MDR 2017/745, list specific glove models covered, include manufacturer details, and be signed by authorized representative. Verify the DoC is dated after May 2021 (MDR application date).
ISO 13485 Certificate: Current quality management system certification from accredited body. Check expiration date and scope to confirm it covers medical glove manufacturing.
Technical File Summary: While full technical files remain confidential, request a summary covering risk analysis, biocompatibility testing (ISO 10993 series), performance specifications (EN 455 for medical gloves), and clinical evaluation.
UDI Registration Proof: Confirmation that glove products are registered in EUDAMED with valid UDI-DI codes. Request the manufacturer’s Single Registration Number (SRN).
Authorized Representative Agreement: For manufacturers outside the EU, verify a valid AR agreement with an EU-established entity responsible for regulatory compliance.
Post-Market Surveillance Plan: Documentation of the supplier’s system for monitoring product performance, handling complaints, and reporting adverse events.
Batch Release Documentation: Certificate of Analysis (CoA) for each production batch confirming compliance with technical specifications and quality standards.
Additional recommended documentation:
- Recent third-party audit reports (if available)
- Material Safety Data Sheets (MSDS) for glove materials
- Biocompatibility test reports (cytotoxicity, sensitization, irritation)
- Performance test results (tensile strength, AQL, viral penetration)
- Supply chain traceability records
- Notified Body certificates (for sterile surgical gloves)
Store these documents in an organized compliance file system with version control. Many distributors now use digital document management platforms that track certificate expiration dates and trigger renewal requests automatically.
Documentation red flags:
- Declaration of Conformity still references MDD instead of MDR
- ISO 13485 certificate expired or issued by non-accredited body
- Supplier unable to provide UDI registration confirmation
- Technical file summary lacks biocompatibility testing references
- No authorized representative listed for non-EU manufacturer
Update your supplier documentation annually at minimum, and immediately if you receive notification of certification changes, product modifications, or regulatory non-conformances.
How Much Does EU MDR Compliance Add to Nitrile Glove Costs
EU MDR compliance typically adds 8-15% to nitrile glove procurement costs through certification fees, enhanced testing requirements, documentation systems, and ongoing surveillance obligations. For a manufacturer producing 100 million gloves annually, initial MDR compliance investments range from $150,000 to $500,000, with annual maintenance costs of $50,000 to $150,000 depending on product portfolio complexity.
Cost breakdown by compliance component:
- ISO 13485 certification: $15,000-$40,000 initial certification, $8,000-$15,000 annual surveillance audits
- Technical file development: $25,000-$100,000 per product family (risk analysis, clinical evaluation, biocompatibility testing)
- UDI implementation: $10,000-$30,000 for system setup, $2,000-$5,000 annual maintenance
- EUDAMED registration: $5,000-$15,000 initial setup, $2,000-$5,000 annual updates
- Authorized representative fees: $8,000-$25,000 annually for non-EU manufacturers
- Enhanced testing: $15,000-$40,000 for comprehensive biocompatibility and performance validation
- Post-market surveillance systems: $20,000-$60,000 for complaint handling and vigilance infrastructure
- Staff training and quality system upgrades: $30,000-$100,000 initial investment
These costs are typically passed through the supply chain, resulting in price increases of $0.50-$2.00 per 100-glove box depending on glove type and manufacturer scale. Larger manufacturers with established quality systems face lower per-unit cost increases than smaller producers who must build compliance infrastructure from scratch.
Cost variables that increase compliance burden:
- Multiple product variants requiring separate technical files
- Sterile surgical gloves requiring Notified Body involvement
- Complex supply chains with multiple raw material suppliers
- Manufacturers new to medical device regulations
- Products requiring extensive clinical evaluation data
For distributors and healthcare facilities, indirect costs include supplier vetting time, documentation management systems, and potential supply disruptions as non-compliant manufacturers exit the market. Many procurement departments report spending 40-60% more time on supplier qualification under MDR compared to previous MDD requirements.
Cost mitigation strategies:
- Consolidate glove purchases with fewer, highly compliant suppliers
- Negotiate multi-year contracts that stabilize pricing despite compliance costs
- Implement digital documentation systems to reduce administrative burden
- Join group purchasing organizations that leverage volume for better pricing
- Consider sterile versus non-sterile glove options based on actual clinical needs
The compliance cost premium is now permanent market reality. Buyers should budget for sustained higher prices rather than expecting costs to decrease as manufacturers complete initial MDR transitions.
Common Mistakes When Implementing Nitrile Glove Traceability Systems
The most common mistake when implementing nitrile glove traceability systems is treating UDI codes as simple label additions rather than integrated data management systems requiring coordination across manufacturing, quality control, and distribution functions. Organizations frequently underestimate the IT infrastructure needed to link UDI codes with production records, batch documentation, and supply chain tracking systems.
Top implementation mistakes:
Incomplete EUDAMED registration: Registering UDI-DI codes but failing to update product information when specifications, packaging, or labeling changes. EUDAMED requires ongoing maintenance, not one-time submission.
Inadequate barcode quality: Using low-resolution printers or incorrect barcode formats that fail scanning validation. GS1-128 and Data Matrix codes must meet ISO/IEC 15416 print quality standards.
Missing UDI-PI components: Including UDI-DI but omitting production identifiers (batch number, expiration date) required for complete traceability. Both components are mandatory on primary packaging.
Disconnected internal systems: Implementing UDI labeling without integrating codes into ERP, quality management, or warehouse management systems. This creates traceability gaps that defeat the regulation’s purpose.
Insufficient staff training: Failing to train production, quality, and logistics personnel on UDI scanning protocols, documentation requirements, and error correction procedures.
Lack of supplier coordination: Not requiring raw material suppliers and contract manufacturers to provide UDI-compatible batch documentation, creating traceability breaks in the supply chain.
Inadequate recall procedures: Implementing UDI codes but not updating recall protocols to leverage batch-level tracking for rapid product retrieval.
Version control failures: Not maintaining historical records of UDI assignments when products are reformulated or repackaged, making it impossible to trace older inventory.
Practical solutions:
- Conduct end-to-end traceability testing before full implementation
- Establish cross-functional UDI governance team spanning quality, IT, and operations
- Implement automated barcode verification at packaging lines
- Create standard operating procedures for UDI data entry and EUDAMED updates
- Schedule quarterly audits of UDI system accuracy and completeness
- Develop supplier requirements documents specifying UDI-compatible documentation formats
A particularly costly mistake is assuming existing lot tracking systems automatically satisfy UDI requirements. While batch numbers provide basic traceability, UDI mandates specific data formats, barcode standards, and database registration that legacy systems often don’t support. Most manufacturers require significant IT investment to achieve full compliance.
For distributors, the common error is accepting supplier claims of “MDR compliance” without verifying UDI codes actually appear in EUDAMED and match product packaging. Always cross-reference supplier-provided UDI-DI codes against the public EUDAMED database before accepting shipments.
Can I Still Sell Non-MDR Compliant Nitrile Gloves in the EU
No, you cannot legally sell non-MDR compliant nitrile gloves as medical devices in the EU after the transition deadlines expired. The final grace period for Class I medical devices (including most examination gloves) ended in May 2024, meaning all medical gloves currently placed on the EU market must meet full MDR requirements including UDI codes, technical documentation, and EUDAMED registration.
Current enforcement status:
- Medical gloves manufactured under old MDD certificates can no longer be placed on the market
- Existing inventory manufactured before May 2024 may be sold until depleted, but this grace period is rapidly closing
- Distributors face legal liability for selling non-compliant devices
- EU member state authorities are actively conducting market surveillance and removing non-compliant products
The regulation distinguishes between “placing on the market” (first making available in the EU) and “making available” (subsequent distribution). Once the transition deadline passed, manufacturers cannot place new non-compliant gloves on the market, though distributors may continue selling existing compliant inventory until it’s exhausted.
Exceptions and alternatives:
You can still sell nitrile gloves in the EU if they are:
- Marketed solely as industrial PPE (not medical devices) under EU Regulation 2016/425
- Labeled for non-medical applications without healthcare claims
- Distributed through non-medical channels with clear “not for medical use” labeling
However, if healthcare facilities purchase these gloves or if marketing materials suggest medical applications, authorities may reclassify them as medical devices requiring MDR compliance regardless of labeling.
Enforcement consequences:
- Product recalls and market withdrawal orders
- Financial penalties up to €1 million or 10% of annual turnover
- Criminal liability for serious violations
- Reputational damage and loss of market access
- Liability for patient harm if non-compliant devices cause injury
Some manufacturers attempted to circumvent MDR by relabeling medical gloves as “industrial” or “general purpose,” but EU authorities have issued guidance clarifying that intended purpose is determined by reasonably foreseeable use, not manufacturer claims. If gloves are predominantly sold to healthcare facilities, they will be regulated as medical devices.
For buyers, purchasing non-compliant gloves creates legal and patient safety risks. Healthcare facilities should verify supplier MDR compliance before accepting deliveries and consider contractual clauses requiring suppliers to indemnify against regulatory non-compliance.
Who Needs to Comply With EU MDR for Nitrile Gloves: Distributors or Manufacturers
Both manufacturers and distributors must comply with EU MDR for nitrile gloves, though their specific obligations differ. Manufacturers bear primary responsibility for device design, testing, technical documentation, and CE marking, while distributors must verify compliance, maintain documentation, and report incidents. EU MDR Article 14 explicitly assigns legal duties to distributors that were largely absent under the old MDD framework.
Manufacturer obligations under EU MDR:
- Design and manufacture gloves according to General Safety and Performance Requirements
- Conduct risk analysis and clinical evaluation
- Create and maintain comprehensive technical documentation
- Implement quality management system (ISO 13485)
- Assign UDI codes and register in EUDAMED
- Issue Declaration of Conformity and affix CE marking
- Establish post-market surveillance and vigilance systems
- Designate authorized representative (if based outside EU)
Distributor obligations under EU MDR:
- Verify manufacturer has issued Declaration of Conformity
- Confirm gloves bear CE marking and required labeling
- Maintain documentation proving supplier compliance
- Ensure storage and transport conditions don’t compromise device integrity
- Report serious incidents and field safety corrective actions
- Cooperate with competent authorities during inspections
- Provide traceability information linking products to manufacturers
- Cease distribution if devices become non-compliant
The regulation introduces the concept of “economic operators” encompassing manufacturers, authorized representatives, importers, and distributors. Each operator in the supply chain shares responsibility for ensuring only compliant devices reach end users.
Distributor liability scenarios:
Distributors assume manufacturer-level obligations if they:
- Rebrand gloves under their own name (private labeling)
- Modify gloves in ways that affect compliance
- Change the intended purpose through marketing claims
- Import gloves from non-EU manufacturers without proper AR designation
Many distributors underestimate their MDR obligations, assuming compliance is solely the manufacturer’s responsibility. However, Article 14 makes clear that distributors who fail to verify compliance or continue distributing non-compliant devices face the same penalties as manufacturers.
Practical compliance approach for distributors:
- Establish supplier qualification process verifying MDR compliance
- Maintain compliance files with DoC, certificates, and UDI documentation
- Implement incoming inspection procedures checking CE marking and labeling
- Create incident reporting procedures for customer complaints
- Train staff on MDR obligations and compliance verification
- Include MDR compliance clauses in supplier contracts
- Conduct periodic supplier audits or reviews
For healthcare facilities purchasing directly from manufacturers, you function as an end user rather than a distributor and have no formal MDR compliance obligations. However, prudent risk management suggests verifying supplier compliance to avoid supply disruptions if non-compliant products are recalled.
How to Audit Nitrile Glove Suppliers for EU MDR Compliance
Auditing nitrile glove suppliers for EU MDR compliance requires a systematic evaluation of quality systems, technical documentation, and regulatory status across three phases: desktop review of compliance documents, on-site assessment of manufacturing and quality processes, and ongoing monitoring of certification status and product performance. Begin with a comprehensive document request covering ISO 13485 certificates, Declarations of Conformity, technical file summaries, and EUDAMED registration proof.
Phase 1: Desktop compliance review
Request and verify the following documents before scheduling on-site visits:
- Current ISO 13485 certificate (check issuing body accreditation and scope)
- EU MDR Declaration of Conformity for specific glove models you purchase
- UDI-DI codes and EUDAMED registration confirmation
- Authorized representative agreement (for non-EU manufacturers)
- Recent Certificate of Analysis (CoA) for production batches
- Post-market surveillance plan and complaint handling procedures
- Summary of technical file including risk analysis and biocompatibility testing
Cross-reference supplier claims against independent verification:
- Verify ISO 13485 certificate authenticity with issuing certification body
- Check UDI-DI codes in public EUDAMED database
- Confirm authorized representative is registered EU entity
- Review any publicly available regulatory warnings or enforcement actions
Phase 2: On-site supplier audit
If desktop review is satisfactory, conduct on-site audits covering:
Quality management system:
- Document control procedures and version management
- Calibration records for testing equipment
- Internal audit schedules and corrective action tracking
- Management review meeting documentation
- Training records for quality-critical personnel
Manufacturing and process controls:
- Raw material qualification and incoming inspection
- In-process quality checks and statistical process control
- Environmental monitoring for contamination control
- Batch release procedures and documentation
- Packaging and labeling verification systems
Traceability systems:
- UDI code generation and assignment procedures
- Batch numbering systems and production records
- Supply chain documentation linking raw materials to finished goods
- Warehouse management and inventory tracking
- Recall simulation and effectiveness verification
Post-market surveillance:
- Customer complaint logging and investigation procedures
- Vigilance reporting systems for serious incidents
- Trend analysis and preventive action processes
- Field safety corrective action procedures
Phase 3: Ongoing monitoring
Establish continuous compliance monitoring:
- Quarterly certificate status checks (ISO 13485, AR agreements)
- Annual review of updated Declarations of Conformity
- Batch-level CoA review for each shipment
- Customer complaint tracking and trend analysis
- Regulatory database monitoring for enforcement actions
- Periodic re-audits (annually for critical suppliers, every 2-3 years for others)
Audit scoring and decision criteria:
Develop a weighted scoring system covering:
- Regulatory compliance (40%): Valid certificates, complete documentation, EUDAMED registration
- Quality systems (30%): ISO 13485 implementation, process controls, corrective actions
- Traceability (20%): UDI systems, batch documentation, recall capabilities
- Post-market surveillance (10%): Complaint handling, vigilance reporting, continuous improvement
Choose suppliers based on:
- Score above 85%: Approved supplier, standard monitoring
- Score 70-85%: Conditional approval, enhanced monitoring, corrective action plan required
- Score below 70%: Not approved, seek alternative suppliers
For smaller organizations without internal audit capabilities, consider hiring third-party quality consultants specializing in medical device supplier audits. Many group purchasing organizations also provide supplier qualification services that members can leverage.
What Happens If My Nitrile Glove Supplier Loses EU MDR Certification
If your nitrile glove supplier loses EU MDR certification, they immediately lose the legal right to place medical gloves on the EU market, and you must cease distribution of their products to avoid regulatory liability. The supplier’s Declaration of Conformity becomes invalid, CE marking is no longer authorized, and continuing to sell their gloves exposes you to enforcement actions including product recalls, financial penalties, and potential criminal liability.
Immediate actions when supplier certification lapses:
Stop accepting new shipments: Do not receive or place orders for products from the non-compliant supplier.
Quarantine existing inventory: Segregate all products from the affected supplier and prevent distribution until compliance status is resolved.
Notify customers: Inform downstream customers and healthcare facilities about the compliance issue and potential need for product retrieval.
Assess regulatory obligations: Determine if you must file incident reports with competent authorities or initiate field safety corrective actions.
Review contractual remedies: Invoke supplier agreement clauses addressing compliance failures, including termination rights and indemnification.
Activate alternative suppliers: Shift orders to backup suppliers with verified MDR compliance to maintain supply continuity.
Certification loss scenarios and responses:
ISO 13485 suspension or withdrawal:
- Supplier cannot demonstrate quality management system compliance
- Existing inventory may be sold if manufactured under valid certification, but no new production
- Supplier must undergo recertification before resuming supply
- Timeline: 3-12 months for recertification depending on non-conformance severity
Authorized representative termination:
- Non-EU manufacturer loses EU-based regulatory representative
- All products become non-compliant immediately
- Supplier must appoint new AR and update EUDAMED registration
- Timeline: 1-3 months if new AR readily available
Declaration of Conformity withdrawal:
- Supplier identifies non-conformance requiring DoC revision
- Existing products may need recall depending on safety impact
- Supplier must resolve non-conformance and issue updated DoC
- Timeline: Variable based on issue severity
EUDAMED registration lapse:
- Supplier fails to maintain current device registration
- Products cannot be legally placed on market
- Relatively quick to resolve if only administrative issue
- Timeline: Days to weeks for database updates
Risk mitigation strategies:
- Maintain relationships with multiple qualified suppliers for critical glove types
- Include certification monitoring and notification clauses in supplier contracts
- Require suppliers to carry regulatory compliance insurance
- Establish safety stock levels that provide buffer during supplier transitions
- Monitor supplier financial health and quality system audit results
- Join industry associations that share supplier compliance intelligence
For healthcare facilities, supplier certification loss can create critical supply shortages, particularly for specialized glove types. Develop contingency plans identifying alternative suppliers for each glove category in your formulary, and maintain strategic inventory reserves for high-use items.
Legal and financial implications:
Distributors who continue selling products from non-compliant suppliers face:
- Joint liability for regulatory violations
- Product recall costs including retrieval, disposal, and customer notification
- Financial penalties from competent authorities
- Litigation risk if defective products cause patient harm
- Reputational damage and loss of customer trust
The best protection is proactive supplier monitoring with automated alerts for certification expiration dates and regular verification of compliance status through EUDAMED and certification body databases.
Are Exam Gloves and Surgical Gloves Treated Differently Under EU MDR
Yes, exam gloves and surgical gloves are treated differently under EU MDR based on their intended purpose, sterility, and risk profile. Surgical gloves are classified as Class I sterile devices requiring Notified Body involvement for conformity assessment, while examination gloves are typically Class I non-sterile devices allowing manufacturer self-certification. This classification difference significantly impacts compliance requirements, costs, and regulatory oversight.
Key regulatory distinctions:
| Aspect | Examination Gloves | Surgical Gloves |
|---|---|---|
| Classification | Class I (non-sterile) | Class I (sterile) |
| Notified Body | Not required | Required for sterility validation |
| Conformity assessment | Manufacturer self-declaration | Notified Body review of sterility |
| Technical documentation | Standard technical file | Enhanced documentation for sterility |
| Performance standards | EN 455 Parts 1-4 | EN 455 Parts 1-4 plus sterility validation |
| Biocompatibility testing | ISO 10993 series | ISO 10993 series plus endotoxin testing |
| Typical cost premium | Baseline | 15-30% higher due to Notified Body fees |
The distinction centers on sterility requirements. Surgical gloves must be supplied sterile for use in invasive procedures, requiring validated sterilization processes, sterile packaging, and Notified Body certification of sterility controls. Examination gloves are supplied non-sterile for diagnostic and protective purposes, allowing simpler manufacturing and self-certification.
Performance requirements for both types:
Both exam and surgical gloves must meet EN 455 standards covering:
- Part 1: Detection of holes (Acceptable Quality Limit testing)
- Part 2: Physical properties (tensile strength, elongation)
- Part 3: Biological evaluation (biocompatibility, protein content)
- Part 4: Shelf life determination
However, surgical gloves face additional scrutiny for:
- Sterility assurance level (SAL) of 10^-6
- Validated sterilization processes (typically gamma irradiation or ethylene oxide)
- Sterile packaging integrity testing
- Endotoxin limits for pyrogenicity
- Enhanced quality control for critical manufacturing parameters
Practical implications for buyers:
When sourcing nitrile surgical gloves versus exam gloves for hospitals, consider:
- Surgical gloves cost 15-30% more due to sterility requirements and Notified Body fees
- Surgical glove suppliers require Notified Body certificates in addition to ISO 13485
- Lead times for surgical gloves may be longer due to sterilization and validation cycles
- Regulatory risk is higher for surgical gloves due to stricter oversight
Some manufacturers produce identical glove formulations in both sterile and non-sterile versions, with the only difference being sterilization and packaging. However, the regulatory pathway and compliance burden differ substantially.
Common misunderstandings:
- “Sterile exam gloves” are classified as surgical gloves under MDR regardless of marketing terminology
- High-quality exam gloves cannot be used for surgery without proper sterility validation
- Surgical gloves used for non-invasive procedures are still regulated as sterile devices
- Powder-free designation applies to both exam and surgical gloves but doesn’t affect classification
For clinical decision-making, use surgical gloves only when sterility is required (invasive procedures, surgery, sterile compounding). Exam gloves are appropriate and more cost-effective for patient examination, diagnostic procedures, and general infection control where sterility isn’t necessary.
Best Practices for Maintaining Nitrile Glove Traceability Records
Best practices for maintaining nitrile glove traceability records require implementing a centralized digital documentation system that links UDI codes to batch production records, supplier documentation, distribution data, and customer delivery information. Establish a minimum 10-year retention policy for all traceability documents and implement automated alerts for certificate expirations, batch recalls, and documentation gaps.
Core traceability record categories:
1. Supplier qualification records:
- ISO 13485 certificates with expiration tracking
- Declarations of Conformity for each glove model
- Technical file summaries and test reports
- Authorized representative agreements
- EUDAMED registration confirmations
- Supplier audit reports and corrective action tracking
2. Product identification records:
- UDI-DI codes for each glove product variant
- UDI-PI data linking batch numbers to production dates
- Product specifications and labeling artwork
- EUDAMED database registration screenshots
- Certificate of Analysis for each received batch
3. Inventory and distribution records:
- Receiving documentation with batch numbers and quantities
- Warehouse location tracking by batch
- Customer delivery records linking batches to specific facilities
- Shipping documentation with UDI-PI information
- Inventory aging reports for expiration management
4. Post-market surveillance records:
- Customer complaint logs with batch traceability
- Adverse event reports and vigilance notifications
- Trend analysis reports identifying quality patterns
- Field safety corrective action documentation
- Recall effectiveness verification records
Digital system implementation:
Modern traceability requires integrated software systems:
- Enterprise Resource Planning (ERP): Core system linking purchasing, inventory, and sales with batch-level tracking
- Quality Management System (QMS): Document control, complaint handling, and corrective action tracking
- Warehouse Management System (WMS): Batch-level inventory location and first-expired-first-out (FEFO) logic
- Supplier Management Platform: Centralized repository for supplier certificates, audits, and compliance documentation
Implement barcode scanning at receiving, storage, and shipping to automatically capture UDI-PI data and eliminate manual data entry errors. Many organizations use GS1-128 barcode scanners integrated with ERP systems to create automatic traceability links.
Record retention requirements:
EU MDR Article 10(8) requires manufacturers to retain technical documentation and post-market surveillance data for at least 10 years after the last device is placed on the market. Distributors should adopt similar retention policies:
- Supplier qualification documents: 10 years after last purchase
- Batch-level traceability records: 10 years after distribution
- Customer complaint records: 10 years after case closure
- Regulatory correspondence: Permanent retention
Audit and verification procedures:
Conduct quarterly traceability audits:
- Select random batches and trace forward to customer deliveries
- Select random customer orders and trace backward to manufacturer batches
- Verify UDI codes in system match physical product labels
- Confirm supplier certificates are current and properly filed
- Test recall simulation by identifying all locations of specific batch within 24 hours
Common record-keeping failures:
- Storing documents in disconnected systems (email, shared drives, paper files)
- Failing to update records when suppliers issue revised Declarations of Conformity
- Not linking customer complaints to specific batches for trend analysis
- Inadequate backup and disaster recovery for electronic records
- Missing documentation for older inventory manufactured under previous certificates
Best practice checklist:
- Implement centralized digital document management system
- Establish automated certificate expiration alerts
- Require barcode scanning at all inventory transactions
- Create standard operating procedures for record creation and retention
- Train staff on traceability documentation requirements
- Conduct quarterly traceability audits and mock recalls
- Maintain secure backup systems with off-site storage
- Establish clear data ownership and access controls
- Document all system changes and version updates
- Perform annual review of traceability system effectiveness
For smaller distributors without sophisticated IT infrastructure, cloud-based quality management systems specifically designed for medical device distributors offer affordable solutions with built-in compliance features, automated workflows, and regulatory reporting capabilities.
Frequently Asked Questions
What is the deadline for EU MDR compliance for nitrile gloves?
The final deadline for Class I medical devices including nitrile examination gloves was May 26, 2024. All medical gloves currently placed on the EU market must meet full MDR requirements including UDI codes and EUDAMED registration. No further extensions are available.
Can I import nitrile gloves from China for EU medical use?
Yes, but the Chinese manufacturer must appoint an EU-based authorized representative, obtain ISO 13485 certification, create MDR-compliant technical documentation, and register products in EUDAMED. The importer must verify all compliance documentation before placing products on the EU market.
How do I verify if a supplier’s UDI codes are legitimate?
Check the public EUDAMED database at ec.europa.eu/tools/eudamed for the manufacturer’s Single Registration Number (SRN) and specific UDI-DI codes. Legitimate codes will appear in the database with complete device information. Request the supplier’s SRN and verify it matches database records.
What’s the difference between UDI-DI and UDI-PI?
UDI-DI (Device Identifier) is a unique code identifying the specific glove model and manufacturer, remaining constant throughout the product’s lifecycle. UDI-PI (Production Identifier) includes variable information like batch number, expiration date, and serial number that changes with each production run.
Do I need different documentation for powder-free versus powdered nitrile gloves?
Yes, each product variant requires separate technical documentation and UDI-DI codes. Powder-free and powdered versions are considered different devices under EU MDR due to different risk profiles and biocompatibility considerations. Note that powdered surgical gloves are banned in many EU markets.
How often should I audit my nitrile glove suppliers?
Conduct comprehensive audits annually for critical suppliers providing high-volume or specialized gloves. Perform desktop compliance reviews quarterly to verify certificates remain current. Increase audit frequency if quality issues, complaints, or regulatory concerns arise.
What happens if I receive gloves without proper UDI codes?
Do not accept delivery or place products into inventory. Contact the supplier immediately to resolve the non-compliance. Accepting and distributing gloves without proper UDI codes violates EU MDR and exposes you to regulatory penalties and recall liability.
Are nitrile gloves for dental use subject to EU MDR?
Yes, nitrile gloves used in dental practices for patient examination or procedures are classified as medical devices requiring full MDR compliance. The intended medical purpose, not the practice setting, determines regulatory classification.
Can I relabel medical nitrile gloves as industrial to avoid MDR requirements?
No, this is regulatory fraud. EU authorities determine device classification based on intended purpose and reasonably foreseeable use, not manufacturer labeling. If gloves are designed for or predominantly used in healthcare settings, they are medical devices regardless of labeling claims.
What’s the cost difference between MDR-compliant and non-compliant nitrile gloves?
MDR-compliant medical gloves typically cost 8-15% more than equivalent non-compliant or industrial gloves due to certification, testing, documentation, and quality system requirements. However, non-compliant gloves cannot legally be sold as medical devices in the EU.
Do I need Notified Body certification for nitrile exam gloves?
No, non-sterile examination gloves are Class I devices allowing manufacturer self-certification without Notified Body involvement. However, sterile surgical gloves require Notified Body certification for sterility validation.
How long does it take a manufacturer to achieve EU MDR compliance?
For manufacturers with existing quality systems, achieving full MDR compliance typically requires 12-24 months. New manufacturers or those without established quality systems may need 24-36 months to implement ISO 13485, develop technical documentation, and complete required testing.
Conclusion
The EU Medical Device Regulation fundamentally transformed nitrile glove procurement, distribution, and compliance management across European healthcare markets. Understanding EU MDR impacts on nitrile gloves, particularly traceability requirements and supplier vetting protocols, is no longer optional for manufacturers, distributors, or healthcare facilities sourcing medical-grade gloves.
The regulation’s core requirements are clear: medical nitrile gloves must carry UDI codes registered in EUDAMED, manufacturers must maintain comprehensive technical documentation and ISO 13485 certification, and distributors share legal responsibility for verifying supplier compliance. The transition deadlines have passed, making full MDR compliance mandatory for all medical gloves currently on the market.
For procurement professionals and supply chain managers, the practical implications require immediate action. Implement systematic supplier vetting processes that verify ISO 13485 certificates, Declarations of Conformity, and EUDAMED registration. Establish digital traceability systems linking UDI codes to batch records and customer deliveries. Conduct regular supplier audits and maintain comprehensive compliance documentation with 10-year retention policies.
The compliance cost premium of 8-15% is now permanent market reality, but the investment protects against regulatory penalties, supply disruptions, and patient safety risks. Organizations that treat MDR compliance as a strategic priority rather than administrative burden will maintain reliable glove supplies while competitors struggle with non-compliant suppliers and enforcement actions.
Next steps for ensuring ongoing compliance:
- Audit your current nitrile glove suppliers against the compliance checklist provided in this guide
- Verify all medical gloves in inventory carry proper UDI codes and are registered in EUDAMED
- Implement quarterly certificate monitoring for ISO 13485 and authorized representative agreements
- Establish relationships with backup suppliers for critical glove categories
- Train procurement and warehouse staff on UDI scanning and traceability documentation
- Conduct mock recall exercises to test your traceability system effectiveness
- Review supplier contracts to include MDR compliance clauses and indemnification provisions
The regulatory landscape will continue evolving as EU authorities refine enforcement approaches and EUDAMED functionality expands. Stay informed through industry associations, regulatory consultants, and official EU guidance documents. Organizations that build robust compliance infrastructure now will adapt more easily to future regulatory changes while maintaining uninterrupted access to quality nitrile gloves for patient care and infection control.
SEO Meta Title: EU MDR Nitrile Gloves: Traceability & Supplier Vetting Guide